Sickle Cell Anemia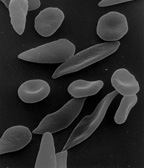
Sickle cell anemia is a genetic disease caused by a mutation in the ß-globin chain of hemoglobin, replacing glutamic acid with less polar valine at the sixth amino acid position. This mutation forms hemoglosin S, which polymerizes in low oxygen conditions, causes them to lose their elasticity, and oxygen-carrying capacity, turning them into sickle-shaped cells (Charache, 1995). Although the cells change back to normal the first time it occurs, with repeated bouts of low oxygen conditions, red blood cells permanently lose their elasticity and become rigid. These sickle-shaped cells travel through the blood stream of the sufferer, becoming trapped and clumped together in narrow pasages, causing extreme pain, ischemia (restriction in blood supply), organ damage, and inevitably, tissue infarction (tissue death) (Charache, 1995).
The sickle cell trait was once considered to be a very advantageous gene, since it prevents the heterozygous carriers of the disease to be immune to malaria in tropical, and subtropical regions, where the Anopheles mosquito inhabits. Only the homozygous recessive individuals, born from two parents who are carriers of the gene, suffer from this very debilitating disorder that was usually fatal before adulthood, prior to several medical discoveries of today (Savitt, 1989)
Since there is no know cure for this condition, the only ways to help
with the pain is through exchange blood transfusions, or with pain medications.
Several complications arising from having sickle cell anemia include,
stroke, gallstones, bone necrosis of the hip, decreased immune reactions,
infarction of penis tissue in men, salmonella osteomyelitis, among others
(Savitt, 1989). 
Starline Hodge: www.starlineprod.com